
生男生女、高龄二胎、失独助孕
单身生育、卵巢衰竭、子宫问题
全球辅助生殖一站式平台19919999145
 全球辅助生殖一站式平台 19919999145
全球辅助生殖一站式平台 19919999145 电话:19919999145
微信:17027022222
邮箱:kf@daiyun.cn
地址:国内、马来西亚、吉尔吉斯斯坦
发布时间:2022-04-15 点此:854次

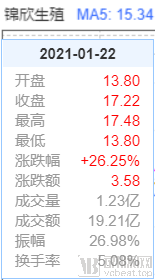
近几日,国内某明星代孕事件一直闹得沸沸扬扬。受到该事件的影响,国内辅助生殖第一股“锦欣生殖”也在事件爆出当天1月18日迎来了猛涨,并连涨3天,截至1月22日,事件已发酵一周,锦欣生殖周涨幅也超过了26%。
代孕并不等同于辅助生殖(ART),辅助生殖只是代孕必须借助的技术手段。在辅助生殖技术加持下,患者夫妇在完成体外受精(IVF)-试管婴儿后,胚胎会被移植回提供卵细胞的母亲子宫内,正常发育生产。而在这个过程中,如果胚胎移植到的是第三方女子的子宫内孕育,就属于代孕行为。人类代孕从技术层面来讲,已经实现完全可操作,但是从法律与道德层面来看,代孕在我国无疑是违法且不被认可的行为。
早在2001年,原卫生部颁布的《人类辅助生殖技术管理办法》,与2003年颁布的《人类辅助生殖技术与人类精子库伦理原则》中就明确指出,医疗机构和医务人员不得实施任何形式的代孕技术。禁止代孕不仅是对弱势妇女、儿童群体利益的保护,同时也是对辅助生殖行业规范化发展的一道警钟。
辅助生殖行业在中国是一个市场化程度与成熟度正在逐步完善的医疗垂直细分赛道,覆盖了治疗服务、医疗耗材、生物医药及遗传检查等多个子领域。该行业不同于其他医疗垂直细分赛道,医药关注的是人类的寿命,而辅助生殖关注的是人类的生育、人类的繁衍。生育问题一直是全人类的话题,背后的人口组成甚至预示了一个国家的未来发展。
近年来,中国不断放宽生育政策,从开放二胎,到解放三胎,这正是社会人口老龄化趋势加剧之下政府采取的应对之策。虽然国家已经从政策层面全面支持国民生育,但是随着现代社会国民生活压力的增大、生活环境及饮食健康等多方面因素的消极影响,民众“不愿生”与“不能生”成为了立在全面放开生育面前的两座大山。“不愿生”需要的是国家各项生育配套与制度改革去解决,而对于“不能生”的核心原因——不孕不育,则需要依靠辅助生殖技术去攻克。也就是说,辅助生殖在我国主要是解决患者夫妇的不孕不育问题,而不是作为代孕或者胚胎性别挑选的工具。
我们将中国近年来对辅助生殖行业的监管进行了简单梳理,以及参考欧美,窥见中国ART的发展未来。
1、中国辅助生殖行业的宏观政策边界
2、辅助生殖上游器械临床豁免加速审批
3、欧美ART行业监管对中国的参考价值
2份管理办法与2份技术规范,划定中国辅助生殖实施边界
我国对辅助生殖行业的系统性规范从2001年颁布首个《人类辅助生殖技术管理办法》开始,在该则管理办法中,首章总则就强调了“禁止以任何形式买卖配子、合子、胚胎。医疗机构和医务人员不得实施任何形式的代孕技术”,为中国的辅助生殖行为划清了基本实施边界。
 中国颁布的辅助生殖法规文件
中国颁布的辅助生殖法规文件
该管理办法自2001年8月1日开始实施,对需要开展辅助生殖的医疗机构需要满足的条件,以及审批流程进行了指示,再对实施过程、违反处罚等做了系统性的说明。同时,《人类精子库管理办法》也在同期颁布,旨在辅佐人类辅助生殖技术安全、有效应用和健康发展。
《人类精子库管理办法》对我国卫生资源、对供精的需求、精子的来源、技术条件等实际情况,制订人类精子库设置规划。人类精子库批准证书每2年校验一次,供精者应当是年龄在22-45周岁之间的健康男性,且只能在一个人类精子库中供精,不得提供超过5名以上妇女受孕等具体指示。
《人类辅助生殖技术管理办法》与《人类精子库管理办法》是奠定我国辅助生殖基础框架的“两大办法”。“两大办法”颁布后,进一步具体的“两大技术规范”也相应出炉——《人类辅助生殖技术规范》与《人类精子库基本标准和技术规范》。
2001年国卫科教颁布“两大技术规范”在2003年经历过一次修改,最终在2003年的修订的《人类精子库基本标准和技术规范》版本中对开展辅助生殖的机构设置条件、在编人员、场地、设备、做出了明确要求,并划线任何生殖机构每年所实施的体外受精与胚胎移植及其衍生技术不得超过1000个取卵周期,并且严禁三胎及以上的妊娠分娩。
同时,该技术规范明文指出:实施技术人员禁止无医学指征的性别选择、禁止实施代孕技术、禁止实施胚胎赠送、禁止以生殖为目的对人类胚胎进行基因操作、禁止人类与异种配子的杂交、禁止开展人类嵌合体胚胎试验研究、禁止克隆人等15项禁止条例,为辅助生殖的操作画出了明确边界。
而另一则《人类精子库基本标准和技术规范》则具体细化了人类精子库的设置条件与管理要求;同年,国家也从伦理方向颁布了《人类辅助生殖技术和人类精子库伦理原则》,奠定了开展辅助生殖的理论基调。
规范之下的中国ART市场:上游器械临床豁免,加速国产品牌落地
在国家出台了一系列宏观辅助生殖管理文件后,中国的辅助生殖行业大致划分为了上游供应器械耗材、检查技术/试剂、生物医药的医疗企业,与下游开展辅助生殖的医疗机构。据统计,目前中国经批准开展人类辅助生殖技术的医疗机构总计517家,经批设置人类精子库的医疗机构总计27家,皆分布于各大省市。
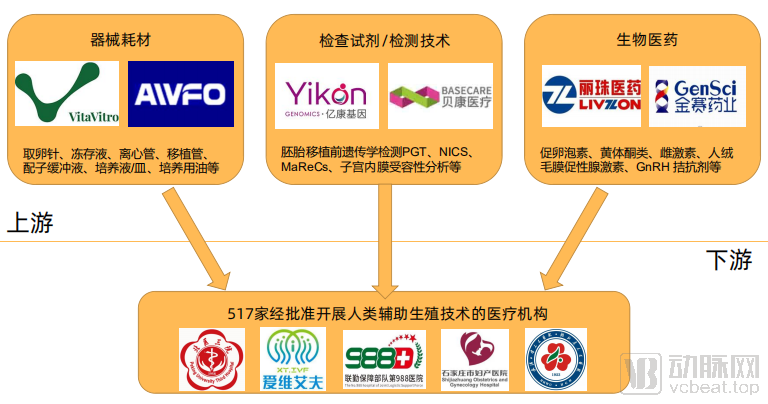
国内辅助生殖上下游布局
上游ART相关的生物医药与普通新药审批流程一致,这里不做过多解读。我们主要关注ART上游的器械耗材的审批,以及遗传检查/检测试剂的审批管理。
器械耗材与检测试剂都属于NMPA的医疗器械审批类目,但他们的审批不同于医药基本已经系统化、流程化(临床前→IND→临床1期2期3期试验→DNA)的审批模式,医疗器械由于不同产品跨度大,依据使用安全性递减顺序分为Ⅰ、Ⅱ、Ⅲ类医疗器械,其中Ⅰ类医疗器械是全面临床的,Ⅱ、Ⅲ类医疗器械主要根据是否纳入NMPA发布的《免于进行临床试验的医疗器械目录》(后简称:免临床目录)中判断,是否需要开展临床试验。但是,医疗器械的临床设计往往相对简单,主要都是针对支持产品上市前的注册临床。
一、中国对ART相关检测试剂的监管
根据《医疗器械分类目录》(2017年第104号),辅助生殖器械分类代码是18-07,其中液体类产品和辅助生育激光系统为Ⅲ类,其他均为Ⅱ类。这里值得一提的是,辅助生殖检测试剂其本质来说是属于基因测序的IVD试剂盒,国家对其的器械分类仍属于Ⅲ类医疗器械。
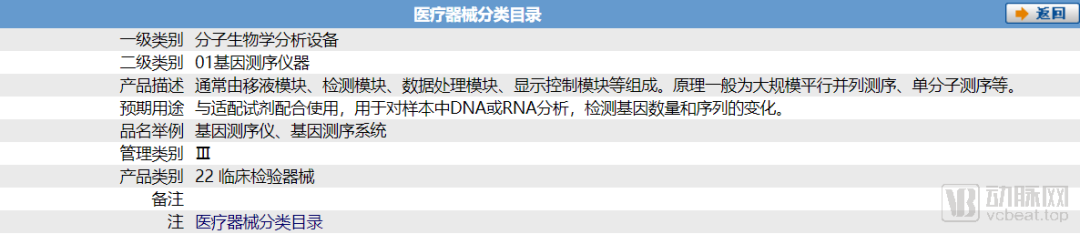
ART相关遗传检测试剂盒被纳入国家Ⅲ类医疗器械目录
而在2020年2月,贝康基因的PGT-A试剂盒(胚胎植入前染色体非整倍体检测试剂盒)就率先获得了NMPA的Ⅲ类医疗器械认证(国械注准:20203400181),成为国内首家获得ART遗传检查试剂批件的医疗公司。试剂盒的获批有助于ART的胚胎植入前染色体非整倍体检测试的大规模应用,也是在同年,国家药监局综合司关于发布YY/T 0506.8—2019《病人、医护人员和器械用手术单、手术衣和洁净服 第8部分:产品专用要求》等24项医疗器械行业标准的公告(2019年第60号)中也将PGT-A试剂盒纳入国家强制性医药行业标准。
随着PGT-A试剂盒的带头,可以预见,未来ART相关遗传检测包括PGS、PGT-SR等也将进一步规范化,纳入国家Ⅲ类医疗器械的审批监管中。
值得一提的是,上游的ART遗传检测解决方案供应商,除了提供IVD试剂盒的商业途径,他们目前还可以采取第三方医学检验的方式为下游辅助生殖中心赋能。例如亿康基因在辅助生殖领域的布局,就是对涉及ART全流程中遗传检测项目的全覆盖,辅助生殖中心的遗传检测需求可以通过与亿康基因这类第三方医学检验合作得以满足。
二、中国对ART相关器械耗材的监管
2001年至2003年间,中国制定的辅助生殖技术与人类精子库相关的管理办法、技术规范、基本标准与伦理原则,成为了后续辅助生殖行业发展的指导标准。同时,国家食品药品监督管理总局(NMPA)关于医疗器械注册申报也出示了一系列指导原则,进一步规范了ART的临床应用。自2015年后,辅助生殖类器械相关的专门法规文件和标准陆续出台,截至目前,已发布了以下指南文件和行业标准:
 国家药监局颁布的辅助生殖类器械相关的法规文件
国家药监局颁布的辅助生殖类器械相关的法规文件
由此可见,国内目前已发布的技术审查指导原则和标准并未覆盖所有的辅助生殖医疗器械产品,特别是行业标准,仅两个Ⅱ类产品(胚胎移植导管和辅助生殖穿刺取卵针)有明确的行标要求。如上文所述,国内辅助生殖类器械的制造商起步也较国外晚,产品成熟度和企业规模不如众多进口品牌,国内注册申报的技巧策略也处于初级阶段,因此,辅助生殖类产品的注册申报和审批过程仍需探索和磨合。
同时,从临床评价路径来看,Ⅱ类产品基本均已纳入免临床目录,Ⅲ类产品也有相当一部分纳入了该目录。这为此类产品的注册提供了极大的便利,不但节省了临床试验的费用和资源,更是为生产企业节约了设计开发中临床确认阶段的时间,加快了企业新产品上市的进程。
“过去因为我国注册难度大,几乎所有ART产品都需要临床试验,因此造成有些技术和产品在我国仍然用的或者说医务工作者已经习惯用的仍然是上一代的。”辅助生殖业内人士告诉动脉网,“但在2019年年底,国家豁免了部分ART产品的注册临床,加速了企业注册进度,让进口和国产品牌都受益。”
目前,国内有一些国产Ⅱ类ART产品的医疗器械注册证,但是国产Ⅲ类液体类产品拿到国内批准的凤毛麟角,且大多是豁免临床之后获批的。这些产品原本的市场基本被进口产品占领,亟须国产企业迎头赶上。值得庆幸的是,成都艾伟孚自主研发的辅助生殖培养用油在2020年8月18日获得NMPA颁发的Ⅲ类医疗器械注册证,成为国内首个获批的辅助生殖培养用油。无独有偶,同样在2021年1月22日,韦拓生物自主研发的玻璃化冷冻液也获得NMPA颁发的Ⅲ类医疗器械注册证,成为国产首个获批的玻璃化冷冻液。
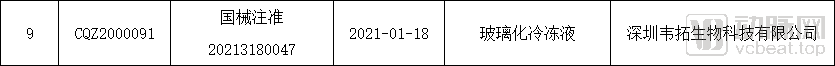
可以预见,随着国家对ART部分器械临床试验的豁免,国内的ART相关器械耗材获批上市进度将会大幅提升。不过这里值得注意的是,医疗器械的临床相对医药显得没那么强制化,这可能导致一些医疗器械公司在法规未强制要求开展临床试验时,很难自主开展各种形式的自发试验,产品虽已上市,但在抢占进口医疗器械市场份额时又会面临一道新的门槛。
这或许除了需要国产品牌认可的市场教育外,还需要医疗器械厂商在自主研发上严于律己、精益求精。韦拓生物创始人林小贞强调,公司此次获批的玻璃化冷冻液虽然国家没有强制要求开展临床(即只需要进行同品种医疗器械临床试验数据对比分析后,便可审批完成注册上市),但公司也本着严谨的态度,也已经自主开展并在中国完成了临床试验;成都艾伟孚创始人严飞也告诉动脉网,该公司自主研发的全程胚胎培养液也在2020年获得国内首个临床试验批件,相信坚持以满足临床真实需求为价值导向的产品,获得临床认可也只是时间问题。
欧美ART监管政策带来的思考
医疗器械标准是医疗器械研制、生产、经营、使用和监督管理共同遵守的技术法规,作为监管的技术支撑,是注册检验和技术审评的依据,是生产质量管理体系的合规性和监督处罚的依据。医疗器械标准还与产业发展密切相关,能规范生产、检验等,降低成本,提高效率,而质量差的标准可能限制产业的发展,甚至引起混乱。
美国食品药品管理局(FDA)是最早关注和实施ART相关医疗器械的安全性评价和监督管理的机构。在美国联邦法规(CFR)的21章中第884部分的G项明确了人类ART用医疗器械的分类和定义,在对该类产品的分类管理中,大多数的种类归为Ⅱ类医疗器械(Class Ⅱ,special controls)与510k同类对比进行管理,该项针对不同类别的产品均提出了框架性的技术要求,如鼠胚试验、内毒素检测、灭菌确认、设计性能规范、生物相容性测试、标签标识要求和临床测试。
在美国的法规中,ART相关医疗器械被归在妇产科器械中,与节育器械等其他的器械分开管理。
 欧美对ART器械的监管法规
欧美对ART器械的监管法规
在欧洲方面,目前欧盟通过医疗器械指令(MDD 93/42/EEC)对无源医疗器械进行管理。
2017年,欧盟官方期刊正式发布了欧盟医疗器械法规(简称:MDR)。MDR于2017年5月26日正式生效,原定经过3年的过渡期在2020年5月26日取代旧的医疗器械指令MDD。但由于受到新冠疫情的影响,欧盟不得不宣布将MDR推迟一年实施,即2021年5月26日。在此之前,无源医疗器械仍在MDD下监管。
在MDD或MDR之外,欧盟于2012年出台了《体外受精和辅助生殖技术产品的合格评价指南》为辅助生殖类医疗器械的合规提供指引。这个指南涵盖了93/42/EEC附录Ⅸ中与IVF和ART相关的医疗器械,从风险管理的角度对ART相关医疗器械进行管理.指南中强调IVF/ART产品的风险和危害程度与其产品的设计生产相关。
欧盟的法规(包括MDD和MDR)强调,对于医疗器械的负效应评估和可接受的风险/受益比的评估,必须立足于充分的临床前评价和临床数据评价,特别强调了产品上市后的临床跟踪。因为辅助生殖不良事件不一定发生在术后,也许会发生在胎儿出生后乃至更久,所以欧盟强调了医疗器械的警戒和临床评价的可追溯性。
目前,MDR尚未正式实施,尽管有些公告机构只接收MDR下的认证申请,但是欧盟尚未发布相关的产品规范(PS,Product Specification),CE认证的策略和流程尚未成熟,尤其是含特别成分的辅助生殖类器械。
综上所述,目前对辅助生殖类产品的监管最成熟的是美国,他们不将此类产品纳入最高监管级别,上市的途径只需经过常规的上市前通告510(k)即可;其次,对辅助生殖类产品的监管最严的是欧盟新法规MDR,只要与配子或胚胎有直接接触,均属于Ⅲ类。这意味着除了液体类产品外,与配子或胚胎接触的针、导管、培养皿之类的在中国被划入Ⅱ类的无源产品也将被MDR划到Ⅲ类中。
相较而言,中国对ART器械的监管要求介于美国和欧盟之间,上市前流程虽没有美国那么简单,但大部分类型产品也没有欧盟那么复杂,Ⅱ类产品较多,Ⅲ类产品也有相当一部分纳入了免临床目录。不过相信,随着国家对ART行业监管的越来越完善,必将促进国产ART品牌的发展;而伴随国家鼓励生育趋势的加剧,辅助生殖赛道也必将成为未来新的一个风口行业。





